Q-Chem Webinar 82
Constrained CASSCF and Tight-Binding Calculations in Q-Chem

Presented by Xinchun Wu on
Xinchun Wu received her B.Sc. in chemistry from the University of Chinese Academy of Sciences in 2020 and is currently pursuing a Ph.D. in Professor Joseph Subotnik’s group at the University of Pennsylvania/Princeton University. Xinchun’s research interests focus on electronic structure method development. She is now working on developing a new electronic structure method called constrained CASSCF (cCASSCF) to capture the charge transfer excited state. Xinchun did an internship in the summer of 2025 at Q-Chem to enable tight-binding calculation and implement the constrained CASSCF method.
Abstract
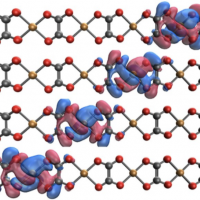
One of the most important and unexplored areas of quantum chemistry is electronic structure in open quantum environments, systems with fractional charges occupying molecular subspaces. To study the electron transfer to and from the bath, one needs an electronic structure method that can give smooth ground and excited potential energy surfaces while avoiding excited states corresponding to internal excitations. For these reasons, our group has developed the constrained CASSCF method. Besides that, the simulation of large open quantum systems requires embedding and approximation strategies, which sparked our idea of approximating the two-electron interaction using a tight-binding model. In the first part of the talk, we will introduce the constrained CASSCF method and how it helps to study nonadiabatic systems, especially in the strong coupling region. We will focus on the motivation for constraining the orbitals, and why this idea of optimizing both charge states is superb for generating smooth and reliable potential energy surfaces and running dynamics. We will also mention how we modified the algorithm in Q-Chem to enable constrained optimization. In the second part of the talk, we will elaborate on adding tight-binding calculation features in Q-Chem. In Q-Chem, by default, all the orbitals that do SCF are calculated ab initio. To extend these calculations to open quantum systems, we need the capacity to work with two different classes of basis functions within Q-Chem – those with and those without two-electron integrals. Moreover, the flexibility to evaluate different parts of the Hamiltonian at different levels of accuracy is crucial. This approach helps bridge ab initio and empirical model calculations.
Supporting Material