Molecular Dynamics
Is your research problem a little bit more dynamic than a purely quantum mechanical approach can handle? Does your enzyme modeling project have some exciting new trajectories in store? If so, Q-Chem's ab initio molecular dynamics (AIMD) and quasi-classical molecular dynamics (QMD) simulations may pique your interest. For larger systems, Q-Chem also has built-in QM/MM and embedding capabilities, as well as interfaces with popular packages like CHARMM and Amber. Discover reaction pathways, predict protein-ligand binding energies, and model the binding of substrates to active sites.
-
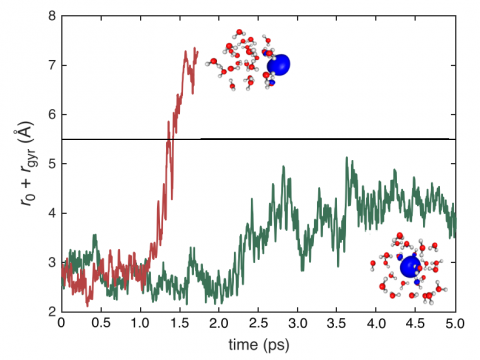
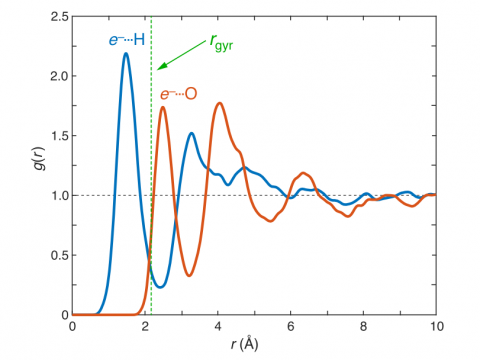
Ab Initio Molecular Dynamics
-
The NVE ensemble (default)
-
The NVT ensemble
-
Langevin Thermostat
-
Nosé-Hoover Thermostat
-
-
Vibrational spectra
-
Quasi-classical molecular dynamics
-
Fewest-switches surface hopping
-
Ab Initio path integrals
-
Want to try Q-Chem?