Explore Q-Chem Features
Q-Chem is a robust software platform with an extensive set of features. Whether you want to study spin-orbit coupling effects in a single-molecule magnet, run high-throughput calculations on small organic molecules, study an enzyme using QM/MM, or something entirely different, our software package offers a wide range of solutions for a variety of applications. Check out our available features and see how Q-Chem can help you achieve your research goals!

Q-Chem 7 is here! Upgrade today and enjoy improved performance and usability, along with a host of new cutting-edge features from Q-Chem developers across the globe.
QC-PBC
QC-PBC is a new module for handling solid-state systems and materials by all-electron calculations with periodic boundary conditions and Gaussian basis sets. Click here for more information!
- DFT and TDDFT
- MP2, LT-MP2, and MP3 for gamma and k-point calculations
- CCSD, CCSD(T), and CCSDT for gamma-point calculations
- Python interfacing
- Geometry optimization
- Solvation
- Analytic frequency and phonon calculations
M-Chem
M-Chem (scheduled to be released later in July) is a new module for handling large biomolecular systems. Learn more here!
- High-performance MPI/OpenMP hybrid implementation for molecular dynamics (MD) simulations
- Fixed charge (Amber), polarizable (AMOEBA), and ReaxFF force fields
- Nose-Hoover thermostat and barostats, conjugate gradient self-consistent field and novel extended Lagrangian schemes for solving the many-body forces, periodic boundary conditions, and particle mesh Ewald
- Modular front-end that stream-lines parameter assignment and system preparation in a more user-friendly and reproducible way for standard force fields and protein-water simulations
- QM/MM integration with ReaxFF
- Trajectory formats for analysis in other codes
Density Functional Theory
- COACH functional: A new range-separated hybrid (RSH) meta-GGA that is more accurate and transferable than the best existing RSH meta-GGAs, such as ωB97M-V, for a wide variety of systems. (Jiashu Liang, Martin Head-Gordon)
- Faster MP2 and double-hybrid DFT: New algorithm provides competitive scaling with DLPNO for MP2 and double-hybrid DFT, with improved error control for higher accuracy. (Zhenling Wang, Yao Shen, Martin Head-Gordon)
- ECD with TDDFT (Xunkun Huang, WanZhen Liang) and EOM-CCSD
- New complex-variable DFT functionals for handling electronically metastable states, including LDA (Slater X, VWN5 C, VWN1RPA C), GGA (B88 X, PBE X, PBE C, LYP C) and hybrid (all hybrid functionals composed of the above LDAs and GGAs and possibly exact HF exchange, including PBE0, B3LYP and BH&HLYP). (Charlotte Titeca, Yifan Jiang, Thomas-C. Jagau)
- Extended tight binding DFT (xtb) energy and gradient (Rebecca Tomann, Siyavash Moradi, Lucas de Kam, Christopher Stein, Martin Head-Gordon)
Correlated Methods
- MRSF-TDDFT with properties: MRSF-TDDFT is an improved version of spin-flip TDDFT that enables effectively spin-pure treatments of doubly excited states, bond-breaking, conical intersections, and some other cases of strongly correlated systems. Q-Chem 7 presents effective MRSF-TDDFT implementation including calculation of state and transition properties, such as state and transition dipole moments, oscillator strengths, spin–orbit couplings, and density-matrix based analyses of the MRSF states and transitions. (Arnab Chakraborty, Zheng Pei, Yihan Shao, Anna I. Krylov)
- New excited-state analysis features based on Earth Mover's Distance (Zhe Wang, Martin Head-Gordon)
- Charge-displacement metrics of TDDFT in libwfa. These metrics are based on are rigorously invariant with respect to orbital rotations, unlike earlier metrics used in other software packages (John Herbert)
- Large speedups in RI-CC2 ground state code in libgmbpt (Hrishikesh Ram and Martin Head-Gordon)
- CC2 and RI-CC2 Dyson orbitals for EA, IP, EE-EA, and EE-IP (Mauro Gascón Navas, Thomas C. Jagau, Robin E. Moorby, Simen Camps, Tianyi Gao)
- THC-sRI Improvements: Includes RI-CC2 and sRI-CC2 oscillator strengths; THC-sRI-CC2 ground and excited state energies and properties; and THC-sRI-CCSD ground and excited state energies (Chongxiao Zhao, Ruihao Bi, Qi Ou, Joohno Lee, Chenyang Li, Wenjie Dou)
- Performance improvements for RI and CD CCSD and EOM-CCSD
- New tools for calculating Auger decay rates using CC/EOM-CC
- NMR chemical shifts improvements (Xiao Liu, Martin Head-Gordon)
- New MPI/OpenMP parallel finite first differences algorithm using RI gives very large speedups over Q-Chem’s legacy NMR code
- Support for all modern density functionals through hybrids
Molecular Dynamics, Non-adiabatic Dynamics, Embedding, and Solvation
- New embedding interface for external packages like CP2K (Ar Fonlon, Elena Kolodzeiski, Dustin R. Broderick, Kay Carter-Fenk, Christopher J. Mundy, Christopher J. Stein, John M. Herbert)
- An order of magnitude total speed up for large QM/EFP jobs via parallelization of one-electron integrals (Lyudmila Slipchenko)
- Pairwise harmonic confiner for optimization in internal coordinates (Chance Brandt, John Herbert)
Fragment and Energy Decomposition Analysis
- Correlated wavefunction EDA: EDA-II is available for MP2, BW-s2, κ-MP2 using the linear-scaling codebase (Zhenling Wang, Hengyuan Shen, Martin Head-Gordon)
- EDA OVOCV analysis and OODFT OVOCV analysis: OVOCV can be used to replace or complement NOCV analysis in EDA; it provides clearer identification of donor and acceptor orbitals than NOCV methods. (Hengyuan Shen, Martin Head-Gordon)
- Two new MBD dispersion models for XSAPT+MBD:
- MBDrev, based on machine learning applied to the SAPT10k data set (Corentin Villot and Ka Un Lao)
- oC8raim, with improved performance for ions and extended to heavy elements (Keegan Paice, John Herbert)
Incorporation of Quantum Nuclear Effects (NEO Suite)
- NEO-AIMD methods for NEO-HF and NEO-DFT, including:
- NEO Born-Oppenheimer MD (NEO-BOMD) (Joseph Dickinson, Sharon Hammes-Schiffer)
- Extended Lagrangian MD (NEO-ELMD) (Joseph Dickinson, Sharon Hammes-Schiffer)
- Constrained NEO MD (CNEO-MD) (Joseph Dickinson, Sharon Hammes-Schiffer)
- NEO-PCM and NEO-QM/MM (Joseph Dickinson, Sharon Hammes-Schiffer)
Click the button below for a detailed list of highlighted features. For a complete list of new features, bugfixes, and improvements, see the Q-Chem 7.0 release log. Interested in trying Q-Chem 7.0 yourself? Request a free demo!

Q-Chem 6.4 was released in December 2025. Highlights in Q-Chem 6.4 include:
- Spectroscopy modeling tools:
- Core-valence separation (CVS) scheme for ROCIS, XCIS, and QROCIS
- Inner-valence projection for EOM-IP/EE-CCSD solvers
- New and improved DeltaSCF driver
- Performance improvements and features for large systems:
- Iterative CC-in-DFT embedding approach for property calculations
- Two-step Cholesky decomposition for CC/EOM-CC energy and gradient
- Stochastic RI-CC2 analytical gradients and derivative coupling.
- DFT and SCF developments:
- Over fifteen new DFT methods
- Support for B97-type functionals in TAO-DFT
- Mixed-reference SF-DFT (MRSF-TDDFT)
- PBEh-3c and HF-3c seminumerical analytic frequency
- NEO developments:
- NEO-CC2 method for excited states
- NEO-CCSD(T), NEO-CCSDTeep,epp methods
- Frozen core approximation for NEO methods
Click the button below for more details on individual features and links to the associated journal publications. For a complete list of new features, bugfixes, and improvements, see the Q-Chem 6.4 release log.

Q-Chem 6.3 was released in May 2025. Highlights in Q-Chem 6.3 version include:
- New analysis tools like the BBOs method and TDDFT charge-transfer metrics;
- Faster DFT performance via optimization of our RI algorithms and MPI parallelization;
- Robust SCF, a sophisticated new "black-box" multi-stage algorithm that selects the optimal SCF algorithm and automatically corrects convergence instabilities, providing improved convergence for SCF and DFT;
- New spectroscopy methods, including several methods for modeling Auger decay;
- New methods for open-shell species, including complex-valued RI-EOM-CCSD, spin-orbit coupling effects in EOM-DEA/DIP, and EOM-DIP/DEA-CCSD gradients;
- Tools for incorporating environmental effects, including heterogeneous PCM ("HetPCM"), density matrix-based and energy-based generalized many-body expansion (GMBE), and extension of SMD solvent frequency calculations to large systems via a semi-numerical frequency implementation;
- Advances in many-body methods for better accuracy, including CC2 with size-consistent Brillouin-Wigner partitioning, and EOM-CCSDT for EE, SF, IP, EA, DIP, and DEA;
- Advances in the NEO suite, including analytic Hessians for fast NEO-PCM calculations, performance improvements, and support for new exchange-correlation functionals;
- Tools for studying chemistry in unusual regimes, including a new mechanochemical pressure model and additional constraints for geometry optimizations and PES scans;
- ...and much, much more!
Click the button below for more details on individual features and links to the associated journal publications. For a complete list of new features, bugfixes, and improvements, see the Q-Chem 6.3 release log.
Q-Cloud (now available!) provides a fast, easy way to run Q-Chem calculations on Amazon's AWS infrastructure, providing improved flexibility and fast turn-around time on jobs while reducing compute costs. The benefits of cloud computing include:
- Flexibility. The number of nodes on your Q-Cloud cluster automatically scales on demand, based on what is actually required to run your jobs. Whether you are running a hundred jobs at once, or just a handful, you'll only pay for the hardware you use.
- Sustainability. According to both Microsoft and AWS, cloud computing options can be 22—93% more energy efficient than traditional on-premises infrastructure, depending on the specific setup.
- Reduced infrastructure costs. Never spend valuable research time troubleshooting faulty hardware again! AWS maintains their own cloud computing infrastructure, so you don't have to, and the Q-Cloud installation process is simple and quick, making it easy to get up and running.
- Fixed-Cost Software-as-a-Service (SaaS) Payment Model. The cost of all standard Q-Cloud licenses is one single monthly or annual payment, and payment for AWS resources scales with use. Additionally, you will always have access to the most recent version of our software.
For more information about the benefits and features of Q-Cloud, you can view our recent webinar, review use cases, or contact our sales team at sales@q-chem.com with questions. Sign up here for a free week-long trial.

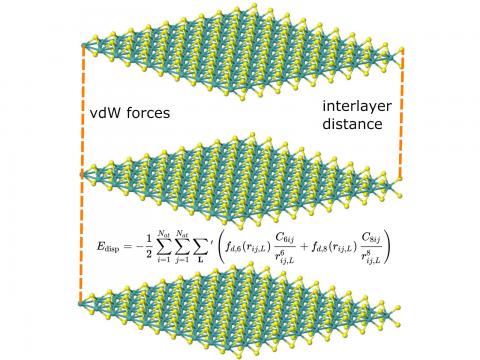
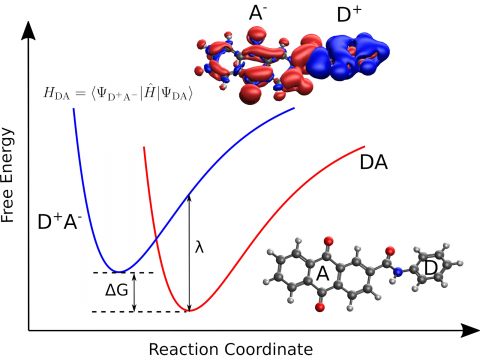
Q-Chem supports LDA, GGA, and meta-GGA functionals, as well as hybrid, range-separated hybrid, and double hybrid versions of both GGAs and meta-GGAs. Single-point energies, geometry optimizations, vibrational frequency calculations, and many other properties can be evaluated for ground states, and for excited states via time-dependent DFT.
Q-Chem offers state-of-the-art tools for treating electron correlation effects, such as Møller-Plesset perturbation theory and coupled-cluster theory. For systems with strong correlation, Q-Chem offers specialty treatments including CASSCF, coupled-cluster valence bond theory, selected CI, RAS-CI, spin-flip, and variational 2-RDM methods.
Q-Chem provides a diverse set of methods for studying electronically excited states: CIS, TD-DFT, NOCI, EOM-CC, and ADC. Specialty flavors of these methods cover many types of electronic structure, making it possible to simulate spectroscopic features, charge and energy transfer, and non-adiabatic dynamics. Additionally, our wavefunction analysis module can be used to provide further insight into excited states.
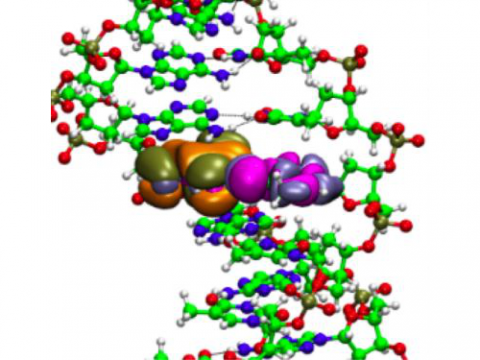
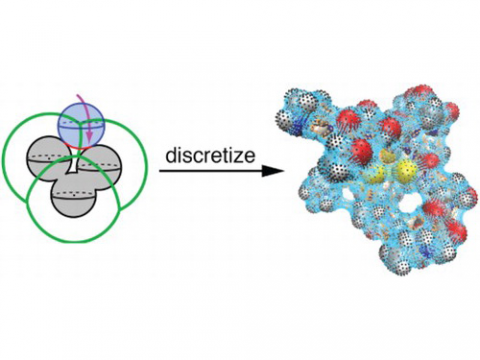
The Q-Chem package offers a variety of solutions for modeling solvated systems, ranging from implicit solvent models, such as SM8, COSMO, and C-PCM, to the effective fragment potential method, which can be used to capture explicit solvent effects. Additionally, Q-Chem includes several different embedding approaches, including QM/MM and density embedding, as well as interfaces to CHARMM and GROMACS.

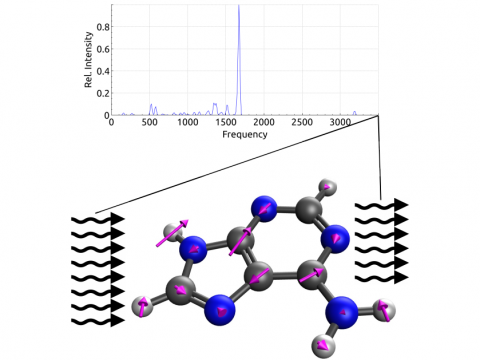
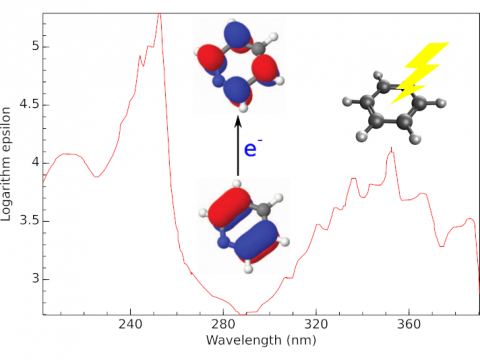
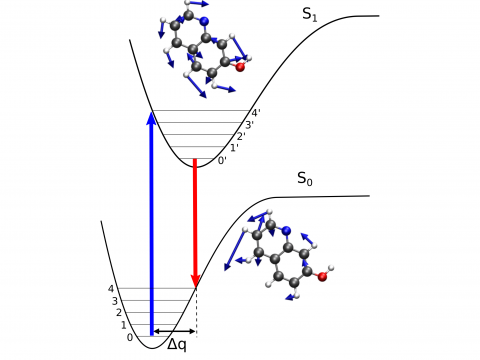
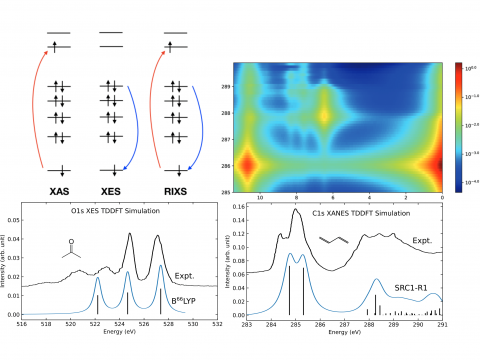
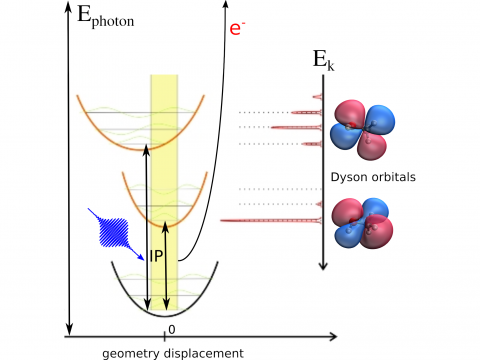
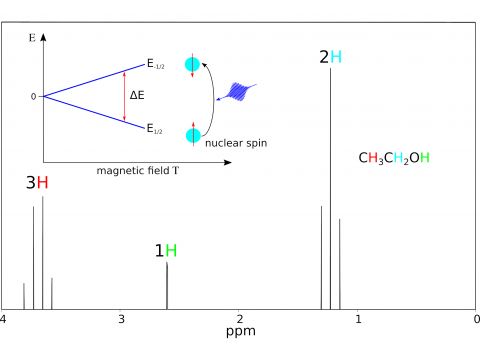
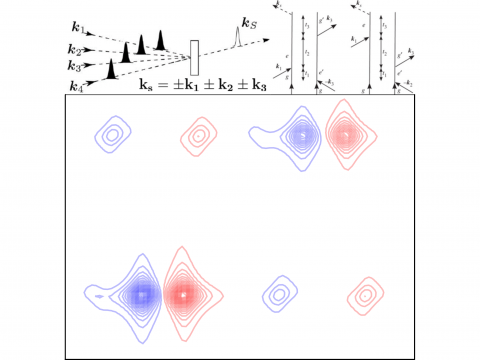
Q-Chem offers a variety of tools for modeling different types of spectra. Our capabilities include IR and Raman spectroscopy, UV-vis spectroscopy, X-ray spectroscopy, photoelectron spectroscopy, NMR spectroscopy, and nonlinear spectroscopy (such as two-photon absorption). Spectroscopic features can be studied using many different levels of theory, ranging from TDDFT to EOM-CC and ADC methods.

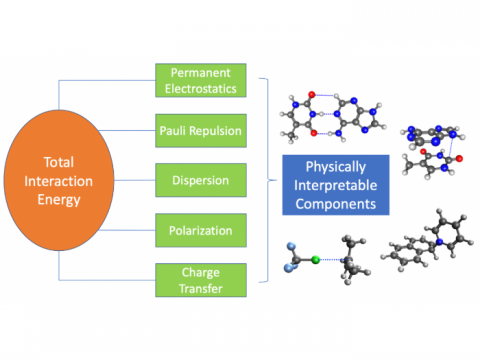
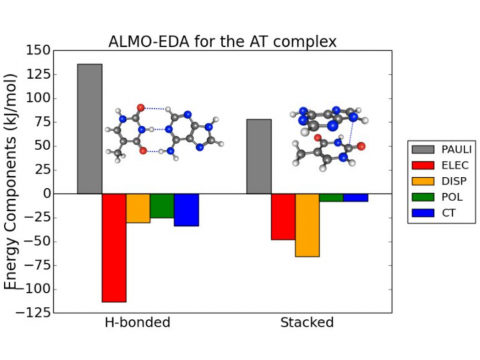
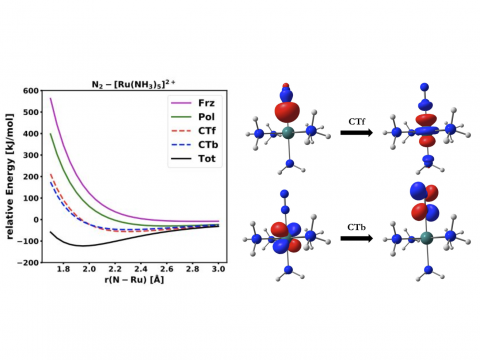
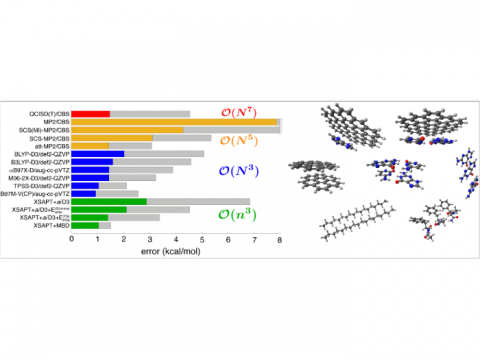
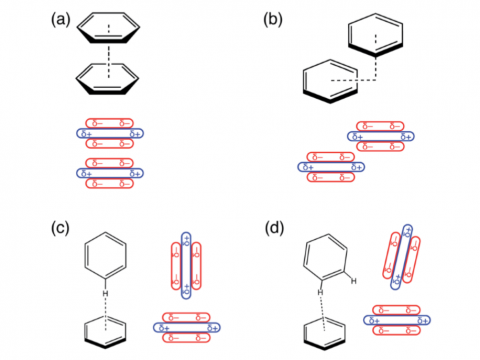
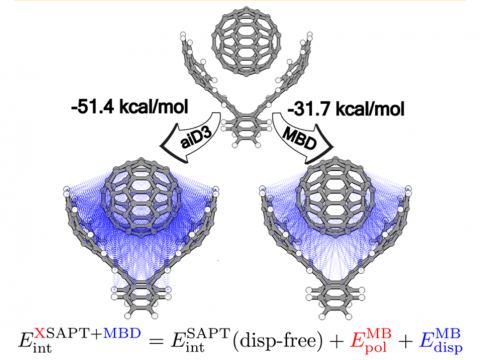
Energy decomposition analysis based on absolutely localized molecular orbitals provides a breakdown of the total interaction energy into meaningful physical terms, providing insights into the nature of intermolecular and bonded interactions. Symmetry-adapted perturbation theory (SAPT) and an extended many-body version thereof (XSAPT) are also available for computing and analyzing intermolecular interactions.

Q-Chem provides methods for geometry optimization, potential energy surface scans, transition state searches, and intrinsic reaction coordinate following, making it ideal for studies of chemical reactivity, thermochemistry, and chemical kinetics.
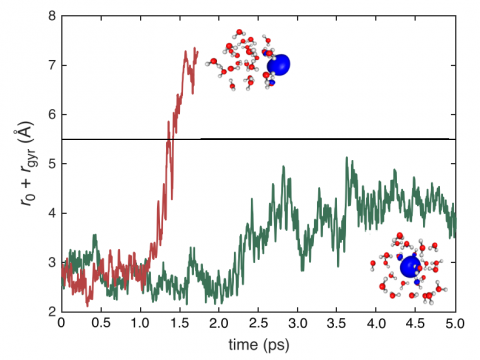
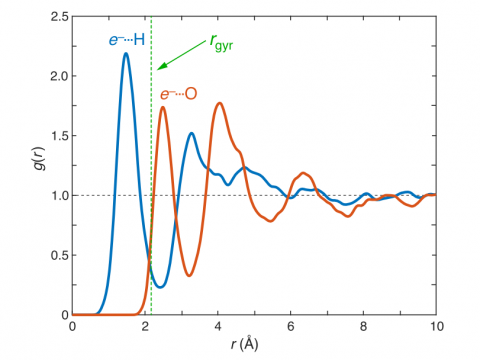
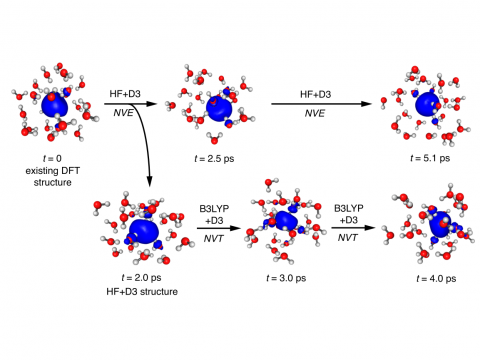
Q-Chem can perform ab initio molecular dynamics (AIMD), including both NVE and NVT thermal samplings, as well as quasi-classical molecular dynamics (QMD). These approaches can be used to produce vibrational spectra and ab initio path integrals. We also include an implementation of Tully's fewest-switches surface hopping (FSSH) approach to effectively handle non-adiabatic systems.