Constrained Density Functional Theory (CDFT)
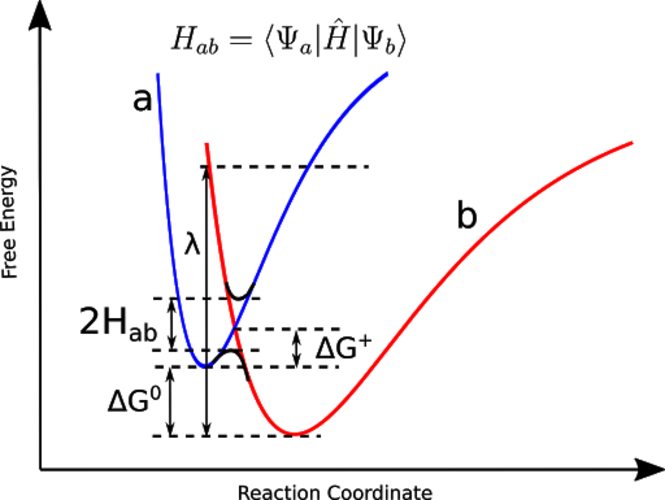
CDFT adds additional potentials to the Kohn-Sham Hamiltonian in DFT calculations in order to obtain charge-localized states, which can be used to approximate diabatic states in electron-transfer reactions.
-
CDFT is a powerful tool to construct diabatic states, which may not be accessible with standard SCF calculations, and to calculate the corresponding electronic coupling and other electron transfer parameters;
-
Q-Chem offers two types of constraints: total charge or spin charge constraints on different molecular fragments.
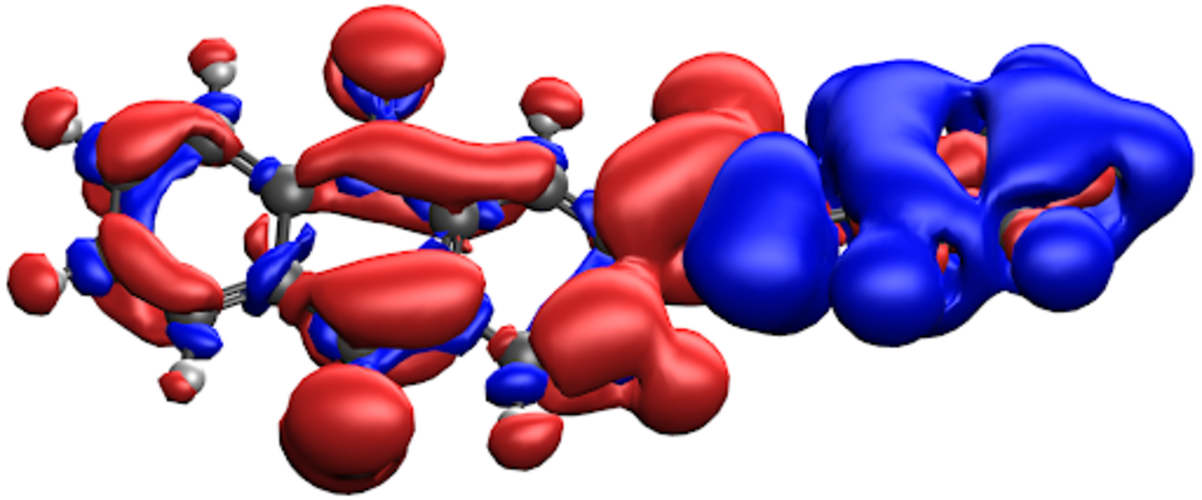 Spin charge of the charge-localized state of the Cu-Ox molecule
Spin charge of the charge-localized state of the Cu-Ox molecule
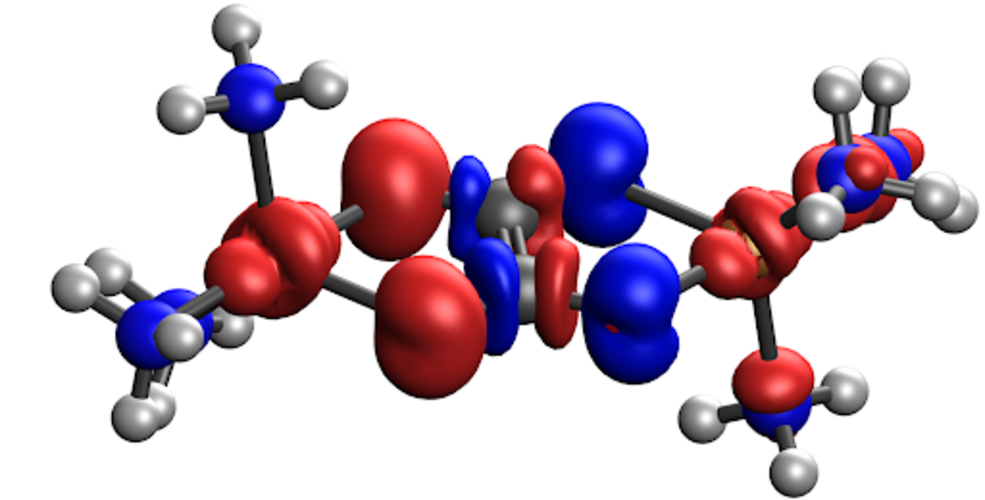 Spin charge of the charge-localized state of the Cu-Ox molecule
Spin charge of the charge-localized state of the Cu-Ox molecule
Predicting Reaction Barrier Heights with Constrained DFT Configuration Interaction (CDFT-CI):
-
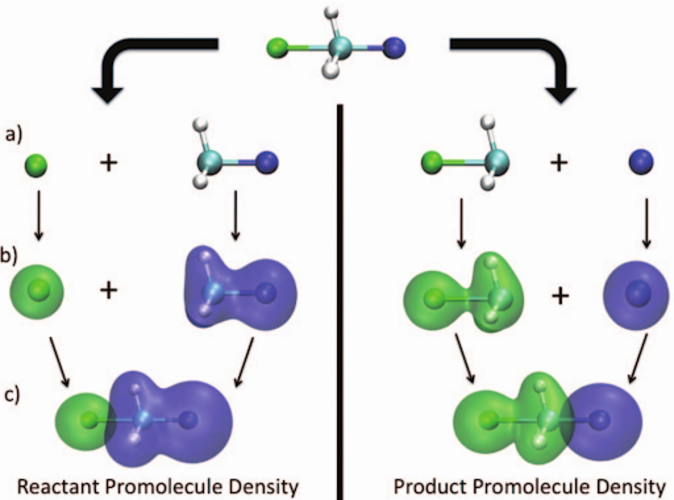
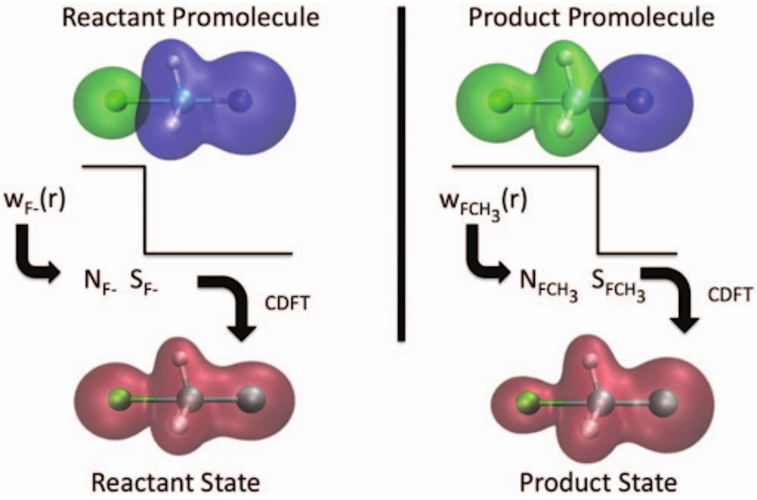
Transition-state energies are searched in the configuration space spanned by two diabatic-like configurations: reactant and product;
-
The reactant and product configurations are obtained by applying charge- and spin-density constraints in DFT calculations, to maximally retain the reactant and product electronic character;
-
CDFT-CI significantly improves the calculated reaction barrier heights compared to traditional DFT.
Want to try Q-Chem?