Q-Chem Webinar 47
Electronic structure methods based on absolutely localized molecular orbitals: from non-covalent interactions to electron transfer

Presented by Dr. Yuezhi Mao on
Yuezhi Mao received his B.S. degree in material chemistry from Peking University in China in 2012. He then joined Prof. Martin Head-Gordon’s group at UC Berkeley and earned his PhD in chemistry in December, 2017. In the Head-Gordon group, he worked on several research projects related to density functional theory calculations and their applications to the study of intermolecular interactions. He is one of the main developers of the second-generation ALMO-EDA method and has extended ALMO-based approaches to the analysis of molecular properties. He is now a postdoctoral scholar in Prof. Tom Markland’s group at Stanford University, and his current research focuses on the study of nonadiabatic processes in the gas or condensed phase using electronic states obtained from ab initio calculations.
Abstract
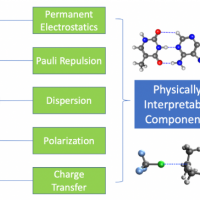
Absolutely localized molecular orbitals (ALMOs) offer a way to construct electronic states with charge localized on molecular fragments. In this seminar, I will introduce the development of electronic structure methods using ALMOs for two distinct objectives. The first is to characterize non-covalent interactions both qualitatively and quantitively, for which we developed the ALMO-based energy decomposition analysis (ALMO-EDA), a scheme that is able to partition a monolithic total interaction energy into physically meaningful, intuitive terms, such as electrostatics, dispersion, etc. I will highlight the recent extension of ALMO-EDA to treat non-covalent interactions in solvation environments, as well as the formulation and application of the “adiabatic” ALMO-EDA that extends the capacity of this method to decompose not only interaction energies but also molecular property shifts arising from intermolecular binding. The second objective is to construct chemically intuitive diabatic states for the study of thermal and photoinduced electron transfer. We obtain variationally optimized charge-localized diabats from density functional theory (DFT) calculations using ALMOs and have developed a modified multistate DFT scheme to accurately predict the electronic couplings between ALMO-based diabats. This approach shows superior accuracy over many other commonly used DFT-based diabatization methods over a broad array of ground-state electron and hole transfer problems. I will then show how one can extend this method to accurately predict diabatic couplings that govern photoinduced electron transfer, by combing the ∆SCF technique with ALMO-based DFT calculations.
[This is an excerpt from our Virtual Wormit Award Mini-Symposium. To view the entire webinar, please visit the Q-Chem YouTube page.]
Supporting Material