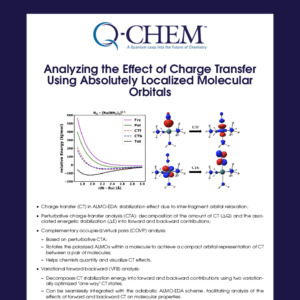
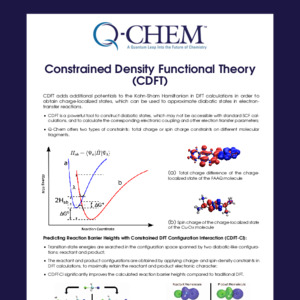
Q-Chem Webinar 41
NEO method available in Q-Chem 5.3: Integrating electronic and nuclear quantum effects

Presented by Zhen (Coraline) Tao on
Zhen (Coraline) Tao is a graduate student in professor Hammes-Schiffer’s group at Yale University. She contributed to the development of electron-proton correlation functionals within the NEO framework. Her current research focuses on studying water clusters with nuclear-electronic orbital density functional theory (NEO-DFT) and NEO dynamics. She joined the Q-Chem developers’ community last year and visited Q-Chem last summer to implement NEO excited state methods within linear response theory.
Abstract
The nuclear-electronic orbital (NEO) approach treats all electrons and specified nuclei, typically hydrogen nuclei, quantum mechanically. This approach can describe nuclear quantum effects such as zero-point energy, nuclear density delocalization, anharmonicity, and vibronic excitations with molecular orbital and density functional theory (DFT) methods. In addition to describing these nuclear quantum effects, the NEO method avoids the Born-Oppenheimer separation between the electrons and quantum nuclei. In this webinar, I will introduce the following methods implemented by Dr. Fabijan Pavošević and myself, which will be available in Q-Chem 5.3: NEO Hartree-Fock (HF), NEO-DFT, NEO excited state methods within linear response theory, such as time-dependent HF and DFT (NEO-TDHF and NEO-TDDFT), and NEO ground state geometry optimizations.* I will also discuss ongoing and future developments within the NEO framework that are being implemented in Q-Chem by the Hammes-Schiffer group.
*F. Pavošević, T. Culpitt and S. Hammes-Schiffer, Chem. Rev. 120, 4222 (2020).
Supporting Material