Q-Chem Newsletter: May 20, 2026
May 20th, 2026
Q-Chem News & Events
Webinar Recording Now Available: OO-DFT Improvements
The recording of our latest webinar from Juan E. Arias Martinez on his recent work creating a new OO-DFT interface in Q-Chem is now available. Watch it here! ⧉
The new driver offers an easy-to-use, streamlined interface, and includes many crucial analysis tools, and he discusses applications of his new interface to studies of organic donor-acceptor charge-transfer excitations.
Q-Chem 7 Pre-Sale
Q-Chem 7 is coming this summer, including many new features, as well as two new packages: QCPBC (for modeling solid-state materials) and M-Chem (for modeling biological systems). Pre-order Q-Chem 7 now to save on licensing fees.
Feature of the Month: Numerical Sparsity Speed-Up
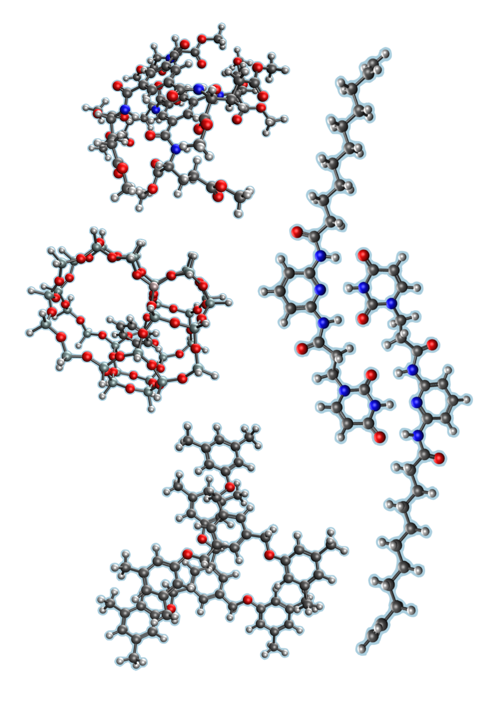
Last year, Q-Chem developers introduced a new numerical sparsity approach to local correlation and linear scaling for MP2. You can read more about it in their JCTC paper here. ⧉
Their method is faster and more accurate than DLPNO-MP2 for the molecules considered in their paper (a subset of which are depicted in the figure on the left). The new approach also includes improved error control for higher accuracy; it only requires setting a single numerical threshold, making it simpler to use.
This new approach will be extended to double-hybrid DFT functionals in Q-Chem 7.
Recent Publication Highlights
Occupied-Virtual Orbitals for Chemical Valence (OVOCV) for EDA and OO-DFT
Occupied-Virtual Orbitals for Chemical Valence with Applications to Charge Transfer in Energy Decomposition Analysis. ⧉ Hengyuan Shen and Martin Head-Gordon. JPC A. 2026.
Researchers recently developed occupied-virtual orbitals for chemical valence (OVOCV). This approach can be used to replace or complement natural orbitals for chemical valence (NOCV) analysis.
The authors recently applied this method to charge transfer in energy decomposition analysis (which you can read about in JPC A here ⧉), where OVOCV provides clearer identification of donor and acceptor orbitals than NOCV methods. They also recently published a preprint on an extension to OO-DFT, which you can read here. ⧉
Excited-State Analysis With MRSF-TDDFT
New Implementation of Mixed-Reference Spin-Flip Time-Dependent Density Functional Theory and Application to Molecules with Variable Diradical Character and Conical Intersections. ⧉ Arnab Chakraborty, Zheng Pei, Yihan Shao, and Anna I. Krylov. Preprint. 2026.
In this recent preprint, authors present a new MRSF-TDDFT implementation in Q-Chem! This includes energies, as well as excited-state analysis via natural orbitals (NOs), natural transition orbitals (NTOs), natural difference orbitals (NDOs), and related descriptors through the libwfa framework.
MRSF-TDDFT is a new method for handling strongly correlated systems that avoids the spin contamination issues common in traditional SF-TDDFT. MRSF-TDDFT energies were added to Q-Chem last year (version 6.4), and we are excited to have this extension to properties included in our upcoming release.
Best Practices for Modeling Reaction Kinetics
Revisiting a large and diverse data set for barrier heights and reaction energies: best practices in density functional theory calculations for chemical kinetics. ⧉ Xiao Liu, Kevin A. Spiekermann, Angiras Menon, William H. Green, and Martin Head-Gordon. PCCP. 2025.
An example of Q-Chem's DFT in action: Last year, authors used Q-Chem for a large benchmark study on RDB7, a large, diverse dataset containing chemical kinetics data for 11,926 reactions. Their work examines common sources of error and establishes best practices for DFT studies of reaction kinetics.
Q-Chem contains a vast array of DFT functionals, including the ωB97X-D3, ωB97M-V, MN15, and ωB97M(2) functionals used in this study.
Additional Publication Highlights
For the most up-to-date paper highlights, follow us on LinkedIn, X, or BlueSky! Want to see your recent paper or preprint featured on our social media posts or in our newsletter? Submit suggestions using our form here!