Q-Chem Newsletter: April 7, 2026
April 7th, 2026
Webinar Recording Available: CC-in-DFT Embedding
Did you miss the recent talk from Anthuan Ferino Pérez on CC-in-DFT embedding? If so, not to worry! The recording is now available on our YouTube channel here. ⧉ Watch to learn more about Anthuan's recent work with projection-based CC-in-DFT embedding in Q-Chem, including exciting practical applications to predicting static polarizabilities of solvated organic molecules.
Upcoming Webinar: OO-DFT Improvements
Join us on April 23rd at 11AM PDT for a talk from Juan E. Arias Martinez on his recent work creating a new OO-DFT interface in Q-Chem! The new driver offers an easy-to-use, streamlined interface, and includes many crucial analysis tools. He will also discuss how he has used this new interface to study organic donor-acceptor charge-transfer excitations. Learn more and register here! ⧉
Feature of the Month: VV10 Analytic Frequencies
Did you know that Q-Chem contains the first published implementation of analytical second derivatives for the VV10 energy? This implementation, developed by Jiashu Liang and introduced in Q-Chem 6, provides faster frequency calculations for several popular DFT functionals like B97M-V, ωB97X-V, and ωB97M-V.
Want to learn more? Read the JCP article from the developers about their implementation here. ⧉
Recent Publication Highlights
COACH Functional: Pushing the Balance of Accuracy, Cost, and Transferability
Reaching for the performance limit of hybrid density functional theory for molecular chemistry. ⧉ Jiashu Liang and Martin Head-Gordon. Preprint. 2026.

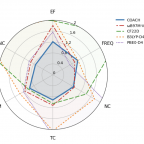
Creating optimal DFT functionals involves a delicate balance between cost, accuracy, and transferability. Researchers Jiashu Liang and Martin Head-Gordon push this balance to the limit with their recently-developed COACH functional. COACH is a new range-separated hybrid (RSH) meta-GGA that is more accurate and transferable than the best existing RSH meta-GGAs, such as ωB97M-V, for a wide variety of systems.
You can read more about COACH in their recent preprint here. ⧉ (The figure on the left, benchmarking COACH's performance against other functionals, is taken from the linked preprint.)
COACH will be available in our upcoming Q-Chem 7.0 release, coming this summer!
Partial Auger Decay Widths from EOM-CC Energies
Computation of Partial Auger Decay Widths from Complex-Valued Equation-of-Motion Coupled-Cluster Energies. ⧉ Florian Matz, Angelos Gkogkos, and Thomas-C. Jagau. J. Phys. Chem. A 2026.
Congratulations to Q-Chem developer Florian Matz, as well as Angelos Gkogkos and Thomas Jagau, on their latest paper in JPC A, which presents an exciting new method for computing partial Auger decay widths faster and more accurately! The new approach they present will be available in a future version of Q-Chem.
How Can We Quantify Electron Transfer?
Is it Possible to Reconcile the Amount of Charge Transfer Defined in Real Space by the Charge Displacement Function and in Hilbert Space by Absolutely Localized Molecular Orbitals? ⧉ Hengyuan Shen and Martin Head-Gordon. JCTC. 2026.
In this recent paper, authors Hengyuan Shen and Martin Head-Gordon explore the question of quantifying electron transfer in hydrogen bonding and other intermolecular interactions. They compare two approaches (CDF in real space, and ALMO-CTA in Hilbert space) to determine why the approaches yield such different results, and discuss how these approaches can be reconciled. They use Q-Chem to compute the intermediate states.
Additional Publication Highlights
For the most up-to-date paper highlights, follow us on LinkedIn ⧉ , X ⧉ , or BlueSky ⧉ ! Want to see your recent paper or preprint featured on our social media posts or in our newsletter? Submit suggestions using our form here! ⧉