Chemical Reactions
Are you exploring a reaction's potential energy surface? Maybe you're interested in exploring some reaction kinetics or calculating a barrier height? Q-Chem provides a suite of tools for calculating and analyzing potential energy surfaces to assist in the understanding of chemical reactions.

Potential Energy Surface Scans
- Both relaxed and unrelaxed PES scans
- Support for scanning over one or two independent geometric variables
- Restrained PES can be used to assist in the search for things like typical SN2 transition states.
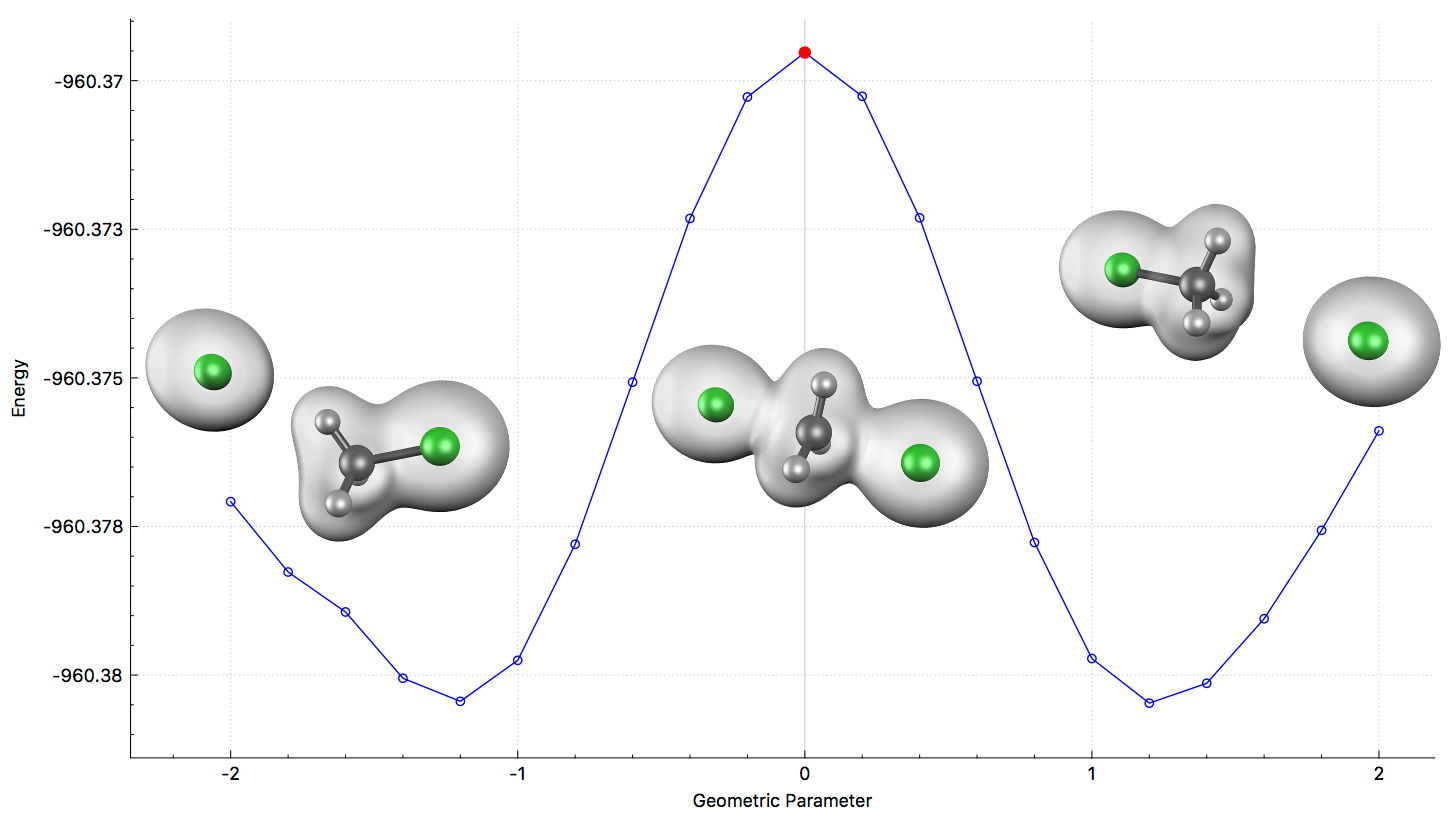

Transition Structure Search
Finding transition structures is critical for accurately predicting kinetic constants and reaction mechanisms. However, transition structures are intrinsically more difficult to obtain than equilibrium structures as they are less intuitive, and the corresponding saddle points are more difficult to locate on a potential energy surface.
- All geometry optimization features are available for transition structure optimization
- The Freezing String Method (FSM) automates the search for candidate transition structures
- Hessian-free methods automatically launched from FSM
- Improved dimer method
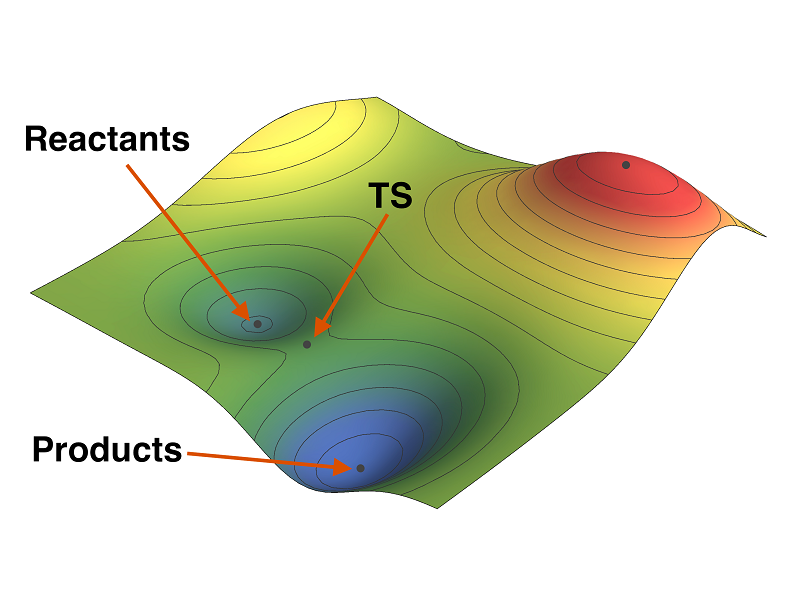

Intrinsic Reaction Coordinate
- Automatically determines which minima are connected to a given transition structure
- Reaction paths can be followed in either Z-matrix, Cartesian or mass-weighted Cartesian coordinates

Minimum Energy Crossing Points
Minimum Energy Crossing Points (MECPs) are essential prerequisites for determining photochemical reaction mechanisms.
- Q-Chem supports several algorithms based on different penalty constraints for locating MECPs.
- MECPs can be optimized at the CIS, SF-CIS, TDDFT, SF-TDDFT, and SOS-CIS(D0) levels of theory.

Freezing String Method
The Freezing String Method (FSM) is an efficient algorithm that automates the search for cadidate transition structures. Like other string methods, it interpolates geometries between reactants and products. Unlike other string methods, the interpolated path is optimized using far fewer gradient calculations, reducing the cost of the search and making the method applicable to much larger systems.


Hessian-Free Methods
Optimising the guess structure using an eigenvector-following algorithm requires the calculation of the Hessian. Q-Chem can avoid this costly step by reusing the output from the FSM to predict the reaction coordinate.
Want to try Q-Chem?

