Simulating X-ray Spectra with Q-Chem
-
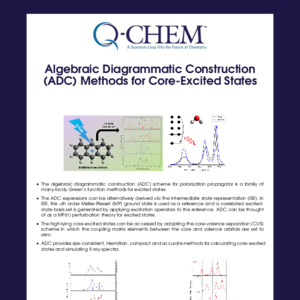
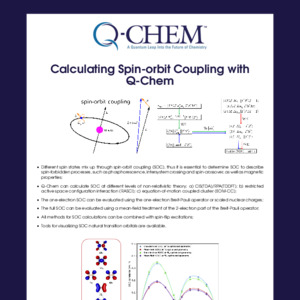
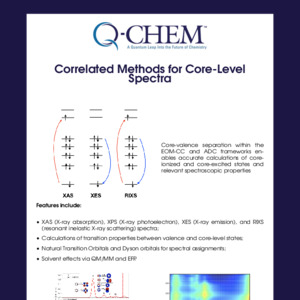
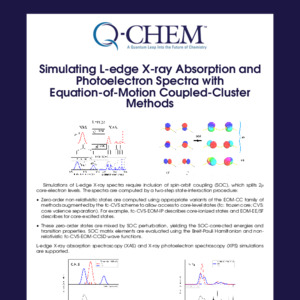
In Q-Chem, the truncated excitation space and core-valence separation approximations are adopted in the time-dependent density functional theory (TDDFT), equation-of-motion coupled cluster (EOM-CC) and algebraic diagrammatic construction (ADC) frameworks, which enables calculations of core-excited and core-ionized states and relevant spectroscopic properties.
-
EOM-CC and ADC are high-level correlated methods which can offer accurate simulated X-ray spectra.
-
TDDFT has a good balance between computational accuracy and cost, making it a widely used tool in X-ray spectroscopy simulation.
-
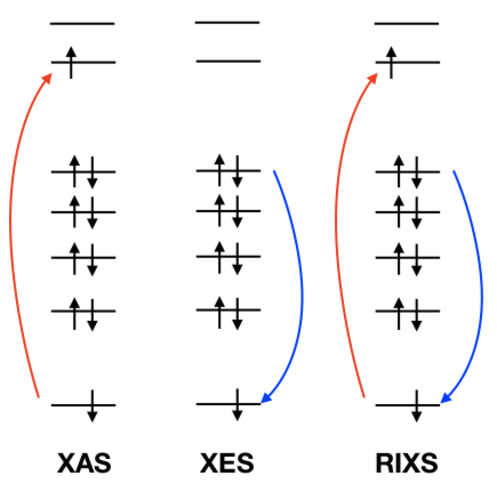
XAS (X-ray absorption), XPS (X-ray photoelectron), XES (X-ray emission) and RIXS (resonant inelastic X-ray scattering) spectra can be simulated.
-
Transition properties between valence and core-level states can be calculated.
-
The simulated spectral features can be analyzed with natural transition orbitals and Dyson orbitals, both of which can be visualized with IQmol.
-
Solvent effects can be included explicitly via QM/MM and effective fragment potential (EFP).
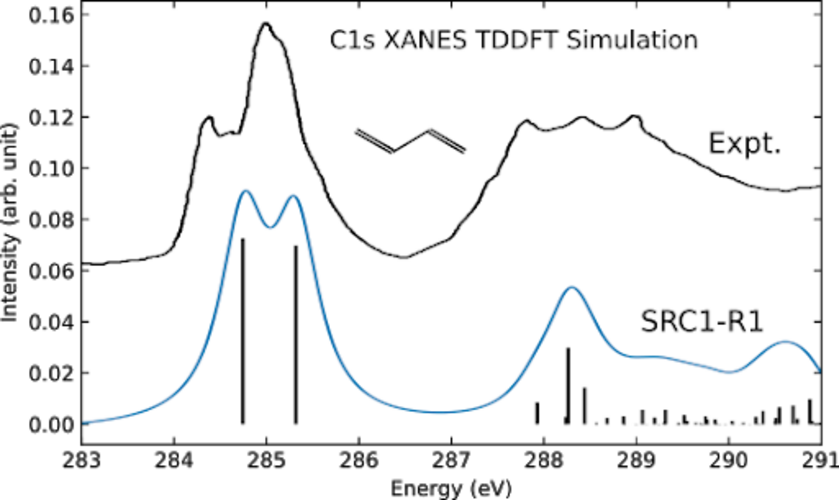 Simulated XAS of butadiene using TDDFT
Simulated XAS of butadiene using TDDFT
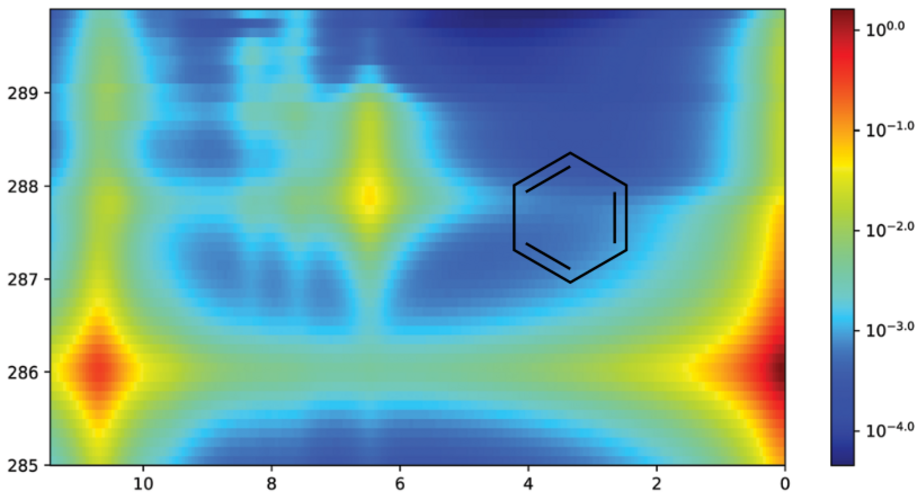 Simulated C K-edge RIXS of benzene using EOM-CC
Simulated C K-edge RIXS of benzene using EOM-CC
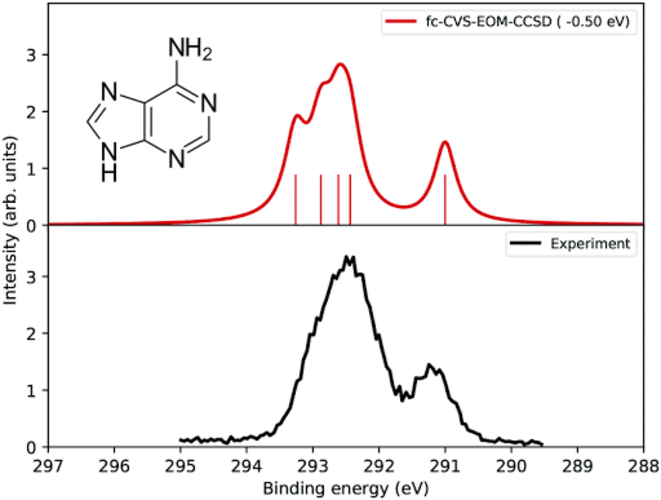 Simulated C K-edge XPS of adenine using EOM-CC
Simulated C K-edge XPS of adenine using EOM-CC
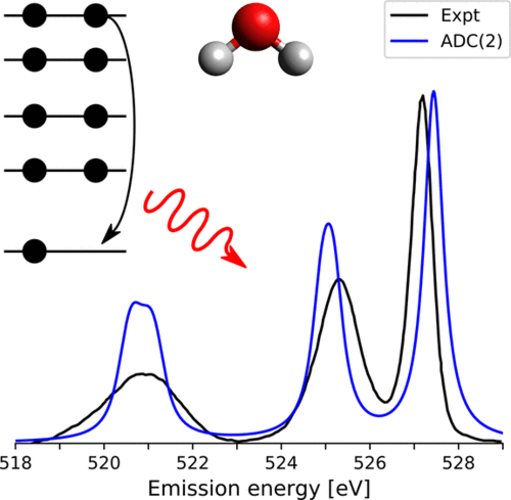 Simulated O K-edge XES of H2O using ADC
Simulated O K-edge XES of H2O using ADC
Want to try Q-Chem?