Single Excitation Theories in Q-Chem
CIS Methods
-
Excited states are computed starting from a Hartree-Fock reference
-
Provides qualitatively correct descriptions of singly excited states
-
Geometries and frequencies comparable to ground-state Hartree-Fock results
-
Efficient, direct algorithm for computing energies, analytic gradients, and second derivatives
-
Perturbative doubles correction via CIS(D) and SOS-CIS(D)
-
Reduces the errors in CIS by a factor of two or more (to roughly that of MP2)
-
RI-CIS(D) and RI-CIS(D0) methods for faster correlated excited-state calculations
-
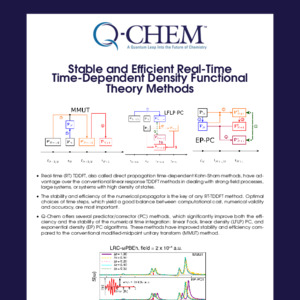
Time-Dependent DFT (TDDFT)
-
Excited-state energies computed from a ground state Kohn-Sham reference
-
Provides a marked improvement over CIS, at about the same cost
-
Captures correlation effects
-
Optimal tuning of range-separated hybrid functionals improves performance for charge-transfer states
-
Spin-flip density functional theory (SF-DFT)
-
Extends TDDFT to states with doubly excited character and conical intersections
-
Useful for bond-breaking, diradicals, and single-molecule magnets
-
Implementation of non-collinear formulation extends SF-TDDFT to a broader set of functionals and improves its accuracy
-
-
Analytic gradients and Hessians of excited states are available
-
Improved accuracy with asymptotically corrected exchange-correlation potential (TDDFT/TDA with LB94)
-
TDDFT within a reduced single-excitation space for special cases
-
Methods for electronic couplings (non-adiabatic and spin-orbit couplings, charge-transfer couplings)