Explore Q-Chem Features
Q-Chem is a robust software platform with an extensive set of features. Whether you want to study spin-orbit coupling effects in a single-molecule magnet, run high-throughput calculations on small organic molecules, study an enzyme using QM/MM, or something entirely different, our software package offers a wide range of solutions for a variety of applications. Check out our available features and see how Q-Chem can help you achieve your research goals!

Q-Chem 6.3 is here! Upgrade today and enjoy improved performance and usability, as well as new tools for studying chemistry and spectroscopy. With these new features, you can deploy large-scale calculations and workflows, get more accurate results faster, and extend the scope of your research to include systems and research questions that were not previously accessible.
Highlights in our latest version include:
- New analysis tools like the BBOs method and TDDFT charge-transfer metrics;
- Faster DFT performance via optimization of our RI algorithms and MPI parallelization;
- Robust SCF, a sophisticated new "black-box" multi-stage algorithm that selects the optimal SCF algorithm and automatically corrects convergence instabilities, providing improved convergence for SCF and DFT;
- New spectroscopy methods, including several methods for modeling Auger decay;
- New methods for open-shell species, including complex-valued RI-EOM-CCSD, spin-orbit coupling effects in EOM-DEA/DIP, and EOM-DIP/DEA-CCSD gradients;
- Tools for incorporating environmental effects, including heterogeneous PCM ("HetPCM"), density matrix-based and energy-based generalized many-body expansion (GMBE), and extension of SMD solvent frequency calculations to large systems via a semi-numerical frequency implementation;
- Advances in many-body methods for better accuracy, including CC2 with size-consistent Brillouin-Wigner partitioning, and EOM-CCSDT for EE, SF, IP, EA, DIP, and DEA;
- Advances in the NEO suite, including analytic Hessians for fast NEO-PCM calculations, performance improvements, and support for new exchange-correlation functionals;
- Tools for studying chemistry in unusual regimes, including a new mechanochemical pressure model and additional constraints for geometry optimizations and PES scans;
- ...and much, much more!
Click the button below for more details on individual features and links to the associated journal publications. For a complete list of new features, bugfixes, and improvements, see the Q-Chem 6.3 release log. Interested in trying Q-Chem 6.3 yourself? Request a free two-month demo!

Q-Cloud (now available!) provides a fast, easy way to run Q-Chem calculations on Amazon's AWS infrastructure, providing improved flexibility and fast turn-around time on jobs while reducing compute costs. The benefits of cloud computing include:
- Flexibility. The number of nodes on your Q-Cloud cluster automatically scales on demand, based on what is actually required to run your jobs. Whether you are running a hundred jobs at once, or just a handful, you'll only pay for the hardware you use.
- Sustainability. According to both Microsoft and AWS, cloud computing options can be 22—93% more energy efficient than traditional on-premises infrastructure, depending on the specific setup.
- Reduced infrastructure costs. Never spend valuable research time troubleshooting faulty hardware again! AWS maintains their own cloud computing infrastructure, so you don't have to, and the Q-Cloud installation process is simple and quick, making it easy to get up and running.
- Fixed-Cost Software-as-a-Service (SaaS) Payment Model. The cost of all standard Q-Cloud licenses is one single monthly or annual payment, and payment for AWS resources scales with use. Additionally, you will always have access to the most recent version of our software.
For more information about the benefits and features of Q-Cloud, you can view our recent webinar, review use cases, or contact our sales team at sales@q-chem.com with questions. Sign up here for a free week-long trial.


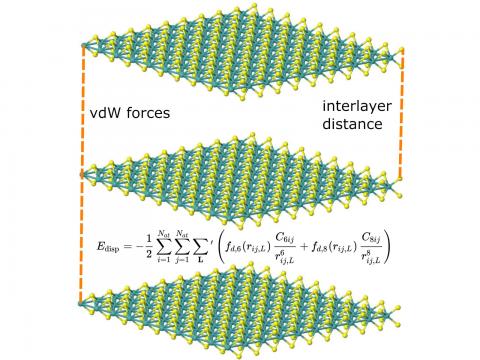
Q-Chem supports LDA, GGA, and meta-GGA functionals, as well as hybrid, range-separated hybrid, and double hybrid versions of both GGAs and meta-GGAs. Single-point energies, geometry optimizations, vibrational frequency calculations, and many other properties can be evaluated for ground states, and for excited states via time-dependent DFT.
Q-Chem offers state-of-the-art tools for treating electron correlation effects, such as Møller-Plesset perturbation theory and coupled-cluster theory. For systems with strong correlation, Q-Chem offers specialty treatments including CASSCF, coupled-cluster valence bond theory, selected CI, RAS-CI, spin-flip, and variational 2-RDM methods.
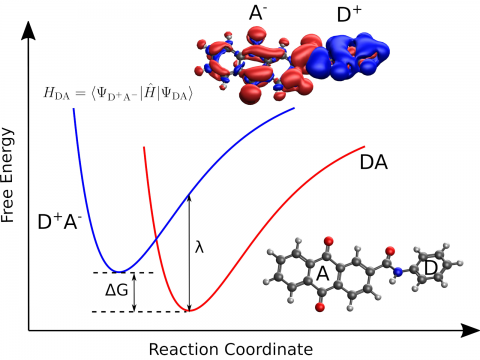
Q-Chem provides a diverse set of methods for studying electronically excited states: CIS, TD-DFT, NOCI, EOM-CC, and ADC. Specialty flavors of these methods cover many types of electronic structure, making it possible to simulate spectroscopic features, charge and energy transfer, and non-adiabatic dynamics. Additionally, our wavefunction analysis module can be used to provide further insight into excited states.

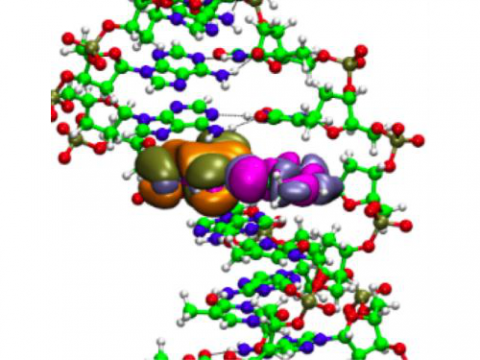
The Q-Chem package offers a variety of solutions for modeling solvated systems, ranging from implicit solvent models, such as SM8, COSMO, and C-PCM, to the effective fragment potential method, which can be used to capture explicit solvent effects. Additionally, Q-Chem includes several different embedding approaches, including QM/MM and density embedding, as well as interfaces to CHARMM and GROMACS.
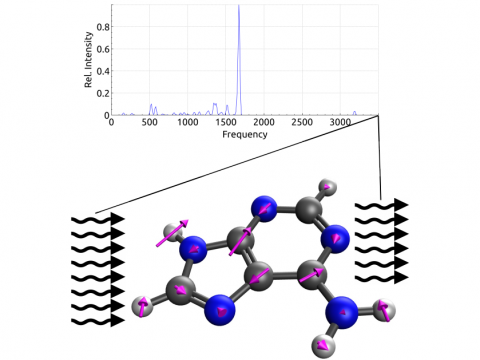
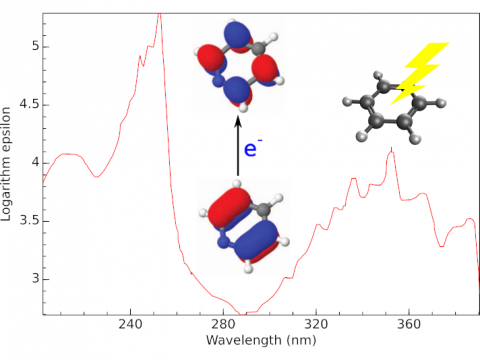
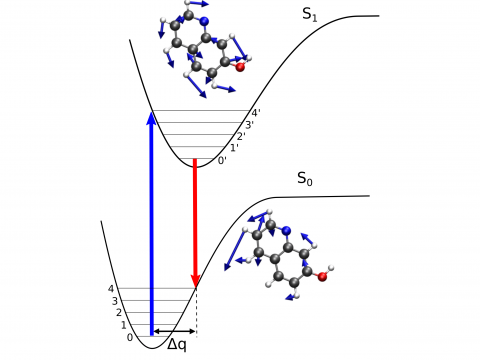
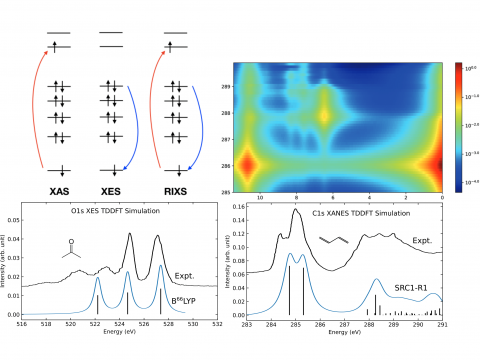
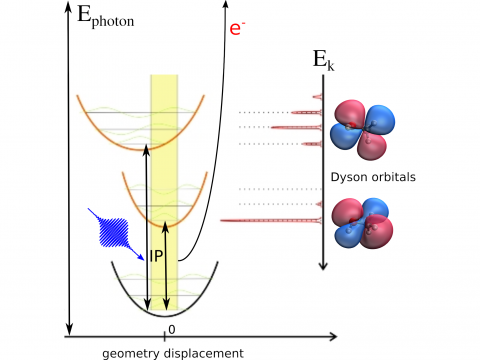
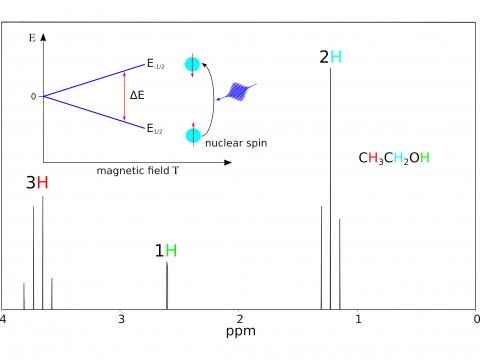
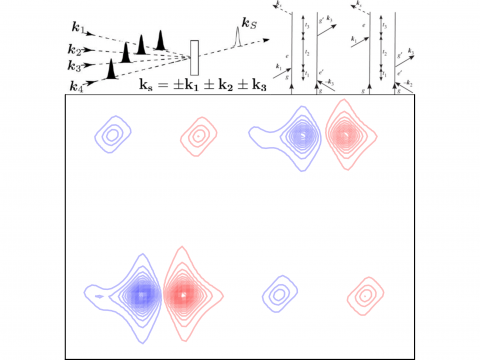
Q-Chem offers a variety of tools for modeling different types of spectra. Our capabilities include IR and Raman spectroscopy, UV-vis spectroscopy, X-ray spectroscopy, photoelectron spectroscopy, NMR spectroscopy, and nonlinear spectroscopy (such as two-photon absorption). Spectroscopic features can be studied using many different levels of theory, ranging from TDDFT to EOM-CC and ADC methods.

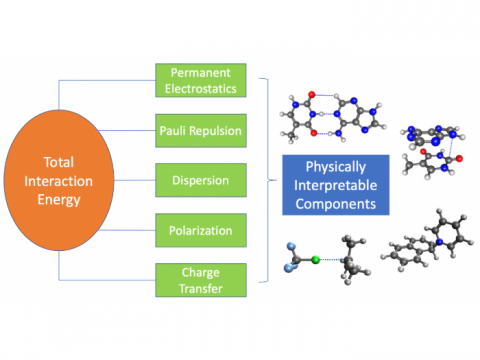
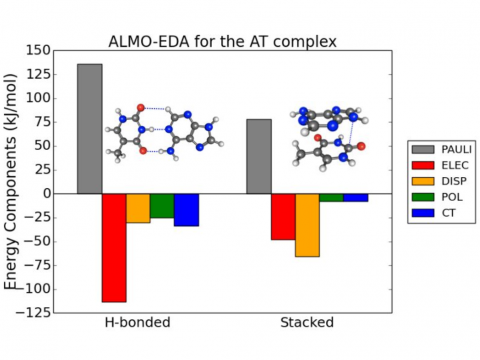
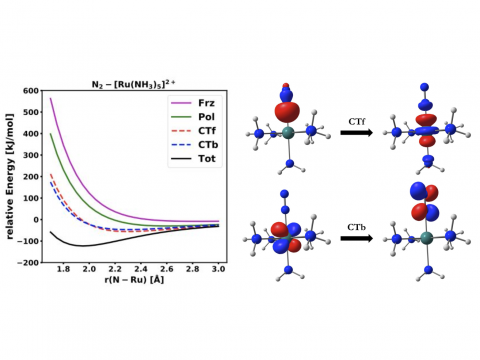
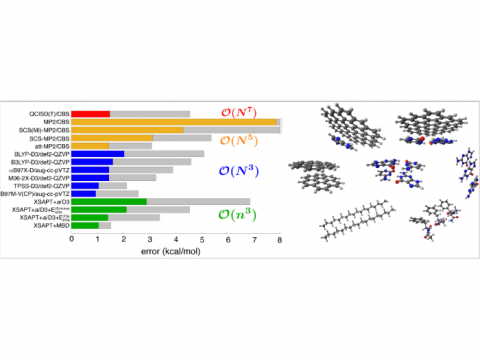
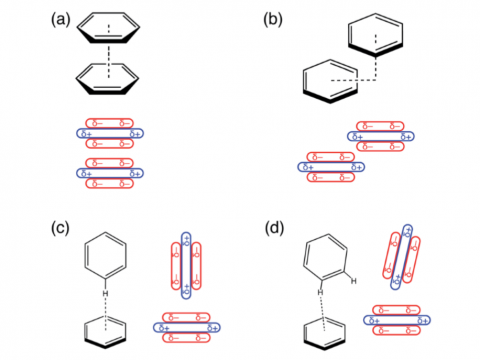
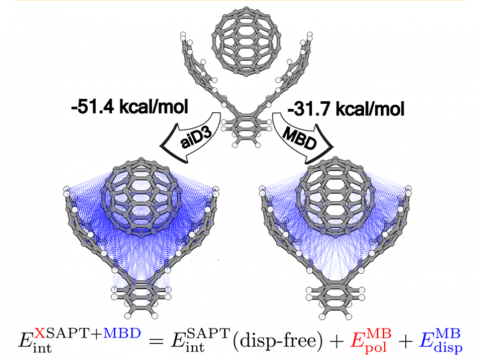
Energy decomposition analysis based on absolutely localized molecular orbitals provides a breakdown of the total interaction energy into meaningful physical terms, providing insights into the nature of intermolecular and bonded interactions. Symmetry-adapted perturbation theory (SAPT) and an extended many-body version thereof (XSAPT) are also available for computing and analyzing intermolecular interactions.
Q-Chem provides methods for geometry optimization, potential energy surface scans, transition state searches, and intrinsic reaction coordinate following, making it ideal for studies of chemical reactivity, thermochemistry, and chemical kinetics.
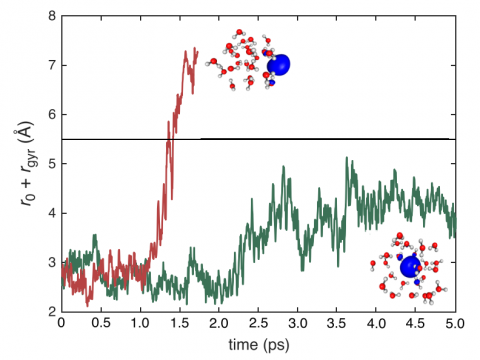
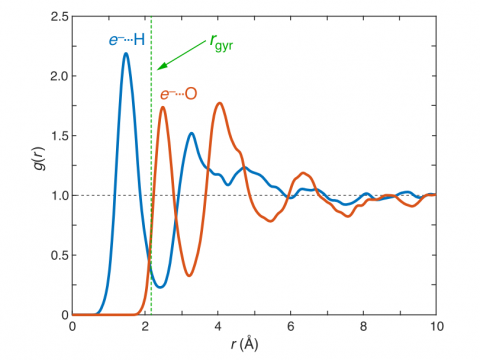
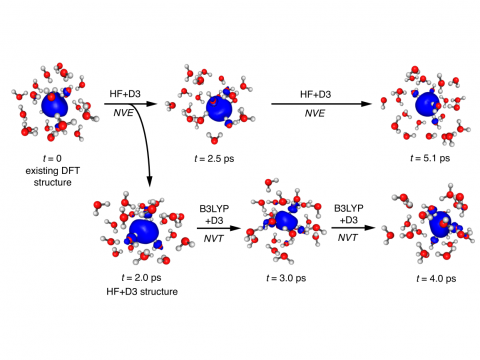
Q-Chem can perform ab initio molecular dynamics (AIMD), including both NVE and NVT thermal samplings, as well as quasi-classical molecular dynamics (QMD). These approaches can be used to produce vibrational spectra and ab initio path integrals. We also include an implementation of Tully's fewest-switches surface hopping (FSSH) approach to effectively handle non-adiabatic systems.